Now that we have mutation load (total, homozygous and heterozygous) estimates for each individual, based on SnpEff and GERP, we can model their effects on lifetime mating success (LMS).
Here, we build three sets of models:
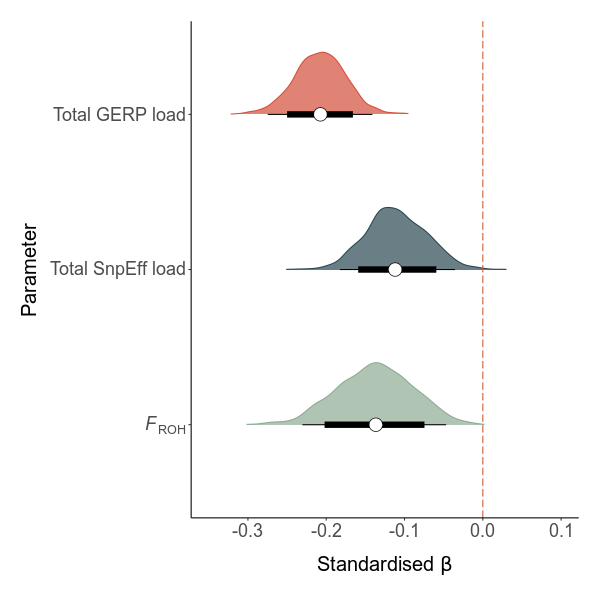
The effect of total load on LMS
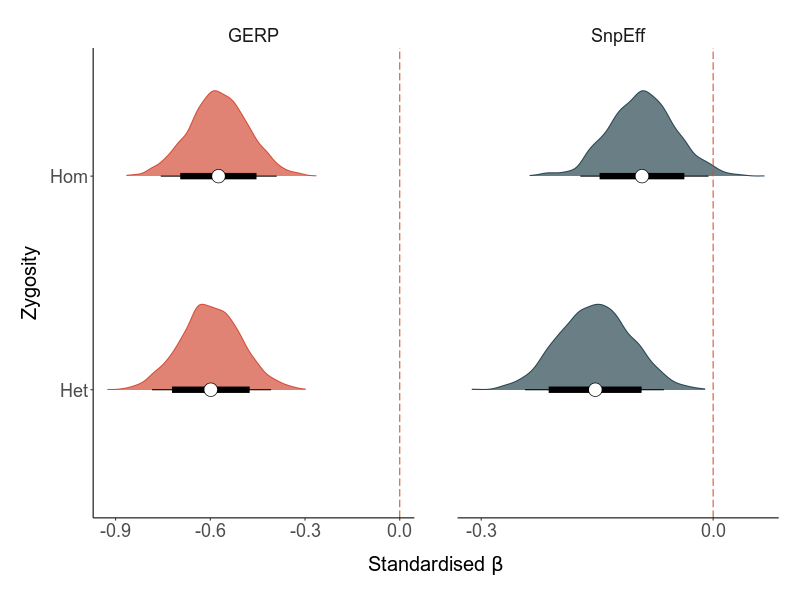
The effects of homo- and heterozygous load on LMS
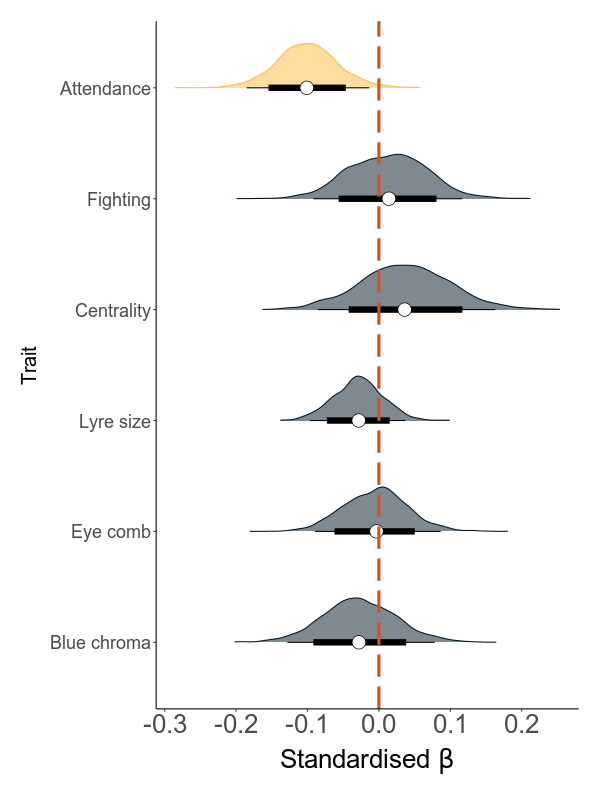
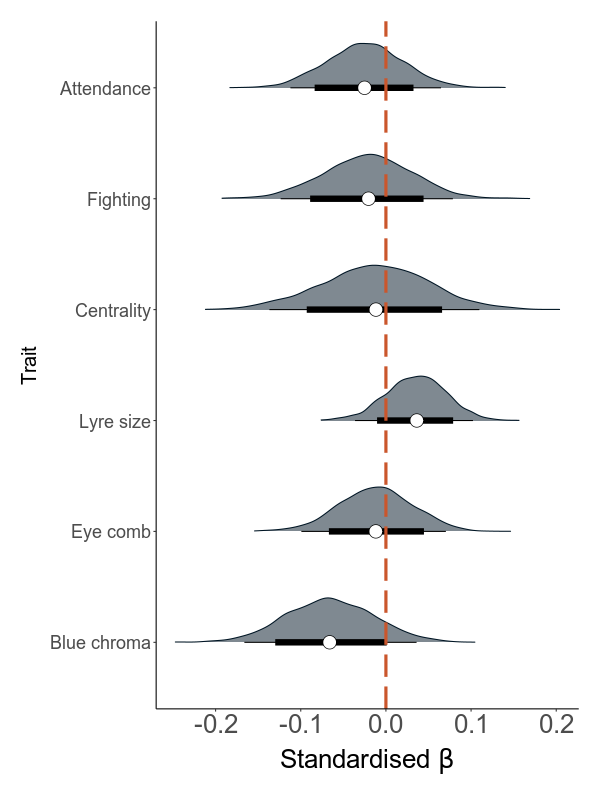
The direct and indirect effects of total load on mating success through the sexual traits
We use Bayesian GLMMs using the R package ‘brms’ to compute these models
5.2 Methods
5.2.1 Total load
The general structure of the total load models is as follows:
LMS ~ scale(total_load) + core + (1|site)
To test for inbreeding depression, we ran a similar model: LMS ~ scale(froh) + core + (1|site)
This is what the script for each load type looks like:
Code
# load pheno data lifetimeload(file ="output/load/pheno_loads_lifetime.RData")# load mutation load measuresload("output/load/all_loads_combined_da_nosex_29scaf_plus_per_region.RData") #loads no sex chr only 30 scaf# subset only the relevant method/loadtypeload <- load_per_region %>%filter(loadtype == method)# combinepheno_wide_load <-left_join(pheno_wide, load, by ="id")pheno_wide_load <-subset(pheno_wide_load, !is.na(total_load)) #some ids without genotypes, excluded for wgr#### model ####brm_load_t <-brm(LMS_min ~scale(total_load) + core + (1|site), data = pheno_wide_load,family ="zero_inflated_poisson",prior =prior(normal(0,1), class = b),cores =8, control =list(adapt_delta =0.99, max_treedepth =15),iter = iter, thin = thin, warmup = warm, seed =1908)save(brm_load_t, file = out)
We can check out the performance of each model using the following loop:
This is what the script for each load type looks like:
Code
# load pheno data lifetimeload(file ="output/load/pheno_loads_lifetime.RData")# load mutation load measuresload("output/load/all_loads_combined_da_nosex_29scaf_plus_per_region.RData") #loads no sex chr only 30 scaf# subset only the relevant method/loadtypeload <- load_per_region %>%filter(loadtype == method)# combinepheno_wide_load <-left_join(pheno_wide, load, by ="id")pheno_wide_load <-subset(pheno_wide_load, !is.na(total_load)) #some ids without genotypes, excluded for wgr#### model ####brm_load_het_hom <-brm(LMS_min ~scale(het_load) +scale(hom_load) + core + (1|site), data = pheno_wide_load,family ="zero_inflated_poisson",prior =prior(normal(0,1), class = b),cores =8, control =list(adapt_delta =0.99, max_treedepth =15),iter = iter, thin = thin, warmup = warm, seed =1908)save(brm_load_het_hom, file = out)
The same diagnostics were applied to each model.
5.2.3 Direct and indirect effects
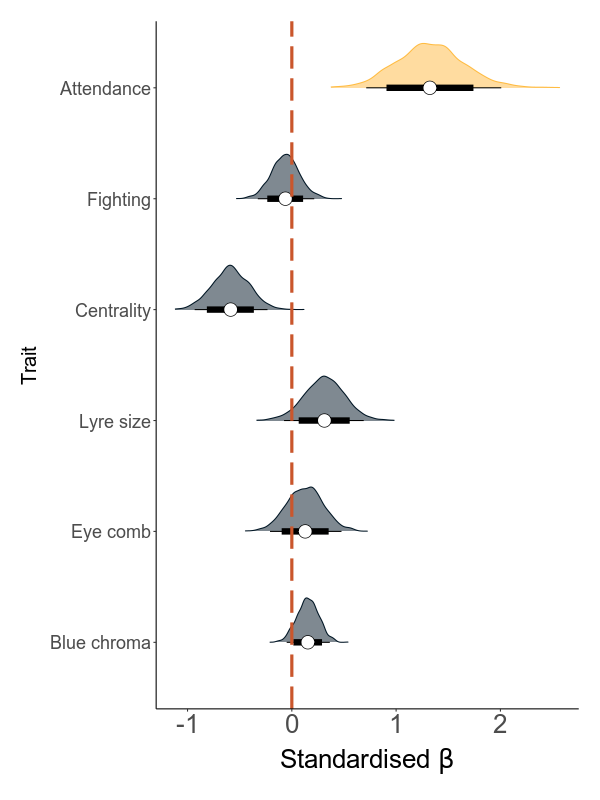
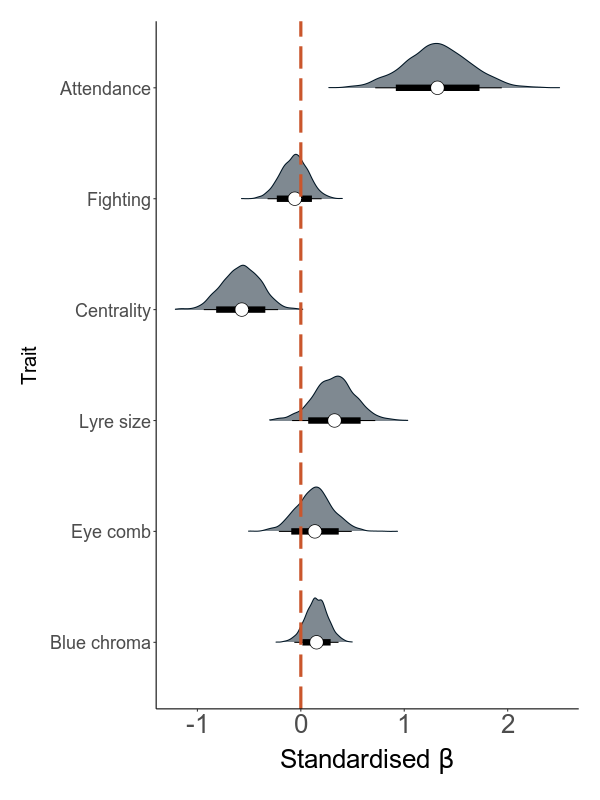
Next, we build models that are based on annual values. There are two sets of models: the first quantifies the effect of load on the six sexual traits (attendance, fighting rate, centrality, lyre size, blue chroma and red eye comb size) in six separate models. The second set analyses the effect of the six traits and load on annual mating success (MS).
The direct effect is the effect of load on MS while correcting for all mediators. The indirect effect is calculated using a mediation analysis, where this effect is calculated as the product of the effect of the predictor (the total load) on the mediator (the sexual trait) and the effect of the mediator on the response variable (AMS).