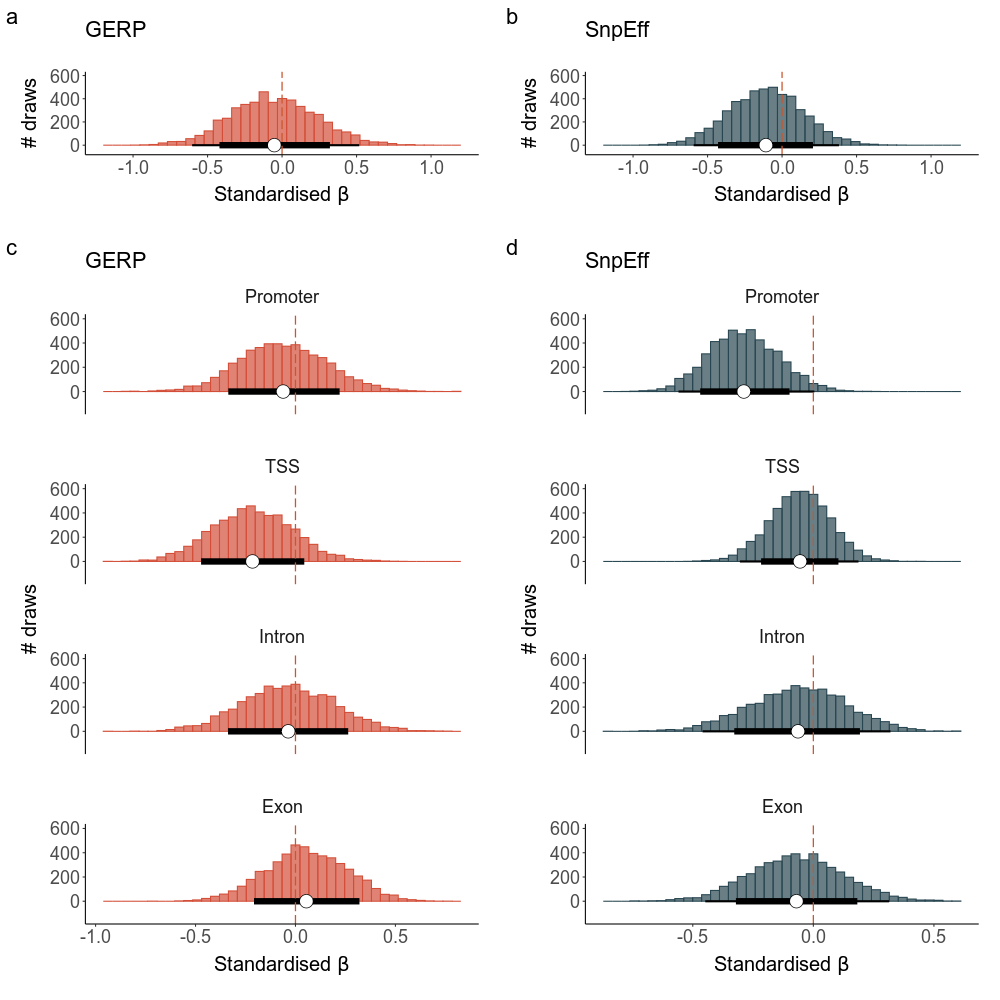
Variation in the effects of mutation load among the various models presented could be attributed to variation in the effect sizes of the individual mutations, or alternatively due to the differences in the number of mutations that contribute to the mutation load estimate. Assuming that all mutations that are identified have negative fitness consequences to some extent, we can expect that a larger number of mutations explain more fitness variation. We tested for the effect of total load on LMS between GERP and SnpEff mutations and the four genomic regions by controlling for the number of mutations.
8.2 Methods
We randomly subset over all deleterioius GERP and SnpEff mutations (separately), and per genomic region for the two approaches.
First, we load in our data and make different subsets of the data: all GERP mutations, GERP mutations per region (4 dataframes) and the same for SnpEff mutations